
Introduction
Degeneration of the intervertebral disc (IVDD) is a leading contributor to lower back pain and is marked by an increase in enzymes that break down the extracellular matrix (such as matrix metalloproteinase) and a surge in the production of pro-inflammatory cytokines. Despite the active efforts of disc cells to preserve the balance of the extracellular environment, their capacity for self-repair is notably insufficient. Beyond the inflammatory reaction in the nucleus pulposus (NP) tissue, the unusual expansion of nerve fibers into the core NP tissue can also significantly alter the internal conditions of the intervertebral disc (IVD) during IVDD development. Our latest review extensively covers the role of the nervous system in the IVD, particularly focusing on how nerve fibers and their neurotransmitters influence IVDD. Moreover, we introduce the concept of ‘neurological IVDD’ for the first time, suggesting that a deeper understanding of how nervous modulation contributes to IVD structural alterations and inflammatory reactions could unveil new therapeutic approaches for managing IVDD.
Previous research highlighted that the neuropeptide, calcitonin gene-related peptide (CGRP), derived from sensory nerves, inhibits cell growth and induces apoptosis, inflammation, and degradation of the extracellular matrix in NP cells. On the other hand, neuropeptide Y, originating from sympathetic nerves, appears to protect NP cells by slowing the degradation process. Vasoactive intestinal peptide (VIP), predominantly found in postganglionic sympathetic nerve fibers, is recognized for its involvement in various conditions, yet its impact on IVDD remains unclear.
Thus, this study focuses on the roles of VIP receptors in human NP tissues and investigates VIP’s potential protective effects against IVDD in laboratory and live models. By doing so, we hope to enhance the understanding of neuromodulation in IVDD and uncover new therapeutic methods.
Results
Decreased Expression of VIP Receptors in Degenerated Human Nucleus Pulposus (NP) Tissue
In our research, we categorized nucleus pulposus (NP) tissue into four different groups, from group II to group IV, based on the Pfirrmann grading scale from MRI findings. Initially, we analyzed the levels of molecules associated with degeneration in these NP tissues. Results demonstrated a gradual decrease in the mRNA levels of ACAN and COL2A1, indicative of cartilage matrix components, in line with increasing severity of NP tissue degeneration. Conversely, we observed a steady increase in the mRNA levels of MMP3/13, which are associated with inflammation and extracellular matrix (ECM) breakdown, as shown in our supplementary figure (Figure S1A). Protein changes corresponding to these genes, as revealed through Western blot analysis, aligned with the mRNA findings (Figure S1A,B in the Supporting Information).
Furthermore, through Safranin O-Fast Green staining and immunohistochemistry (IHC), we found diminished glycosaminoglycan content in degenerated NP tissues. This reduction was directly related to the degree of degeneration, as the presence of MMP3-positive NP cells increased with higher Pfirrmann scores (shown in Figure 1A,B).
Following the investigation of degenerative markers, we assessed changes in the expression of VIP receptors (VIPR1 and VIPR2) in the NP tissues. IHC staining revealed a decline in both VIPR1 and VIPR2 levels as degeneration worsened, indicating a dose-dependent relationship with tissue degradation (also illustrated in Figure 1A,B).
To corroborate these observations, we established an in vitro model of NP cell degeneration using interleukin-1 beta (IL-1β) and hydrogen peroxide (H2O2). In this model, both IL-1β and H2O2 led to a dose-dependent decrease in the expression of VIPR1 and VIPR2 in human NP cells, reinforcing our previous findings.
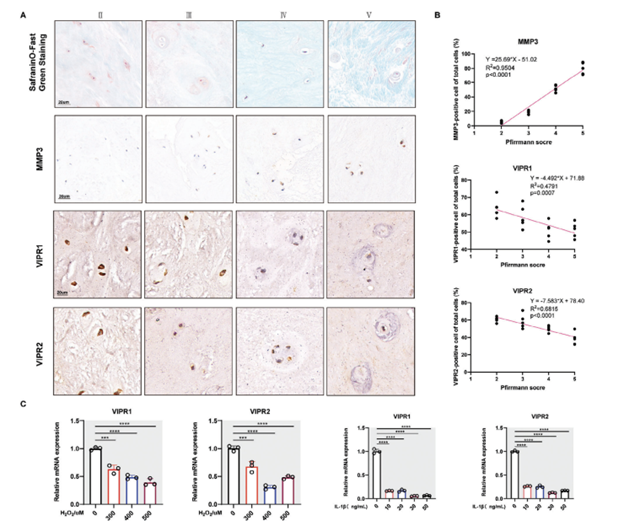
Figure 1. The expression of VIP receptors was decreased in human degenerated NP tissue. A) Representative images regarding SOFG staining and, IHC staining of human NP tissue of patients with different degrees of degeneration according to the Pfirrmann MRI grading system. B) Linear regression of the expression of MMP3, VIPR1, and VIPR2 in human NP tissue of patients with different degrees of degeneration according to the Pfirrmann MRI grading system. C) qRT-PCR results of VIPR1 and VIPR2 in vitro human NP CELLS degeneration treated by IL-1B and H2O2 in a dose-dependent manner. All data are shown as the mean ± SD. Significance was calculated by Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
VIP Regulated Human NP Cells Degeneration Mainly via VIPR2
Figure 2 displays that NP cells extracted from grade II human NP tissue showed significant levels of VIPR1 and VIPR2 expression (Figure 2A). To investigate the functions of these receptors in NP cells, we selected si-RNAs with optimal transfection efficiency targeting VIPR1 and VIPR2. The sequences used were for VIPR1: sense 5′-GUCAUGGCUAACUUCUUCUTT-3′ and antisense
5′-AGAAGAAGUUAGCCAUGACTT-3′; and for VIPR2: sense 5′-GAGCUUCUGAGGUCUCAAATT-3′ and antisense 5′-UUUGAGACCUCAGAAGCUCTT-3′ (Figure 2B). According to the qRT-PCR results, while VIPR1 knockdown did not significantly impact the genes, suppression of VIPR2 notably reduced the expression of ACAN and COL2A1 and increased MMP3/13 levels (Figure 2C).
Further analysis involved bulk transcriptome sequencing of human NP cells treated with IL-1β in the presence or absence of VIP. The bioinformatic evaluation showed that an inflammatory stimulus reduced VIPR2 expression, while administration of VIP to NP cells led to an upregulation of VIPR2 (Figure 2D). Thus, we inferred that VIPR2 likely plays a crucial role in mediating VIP’s regulatory effects on IVDD.
Western blot analysis corroborated these findings by demonstrating that VIP could partially counteract the degenerative effects of IL-1β on human NP cells in vitro, predominantly through the involvement of VIPR2 (Figures 2E,F).
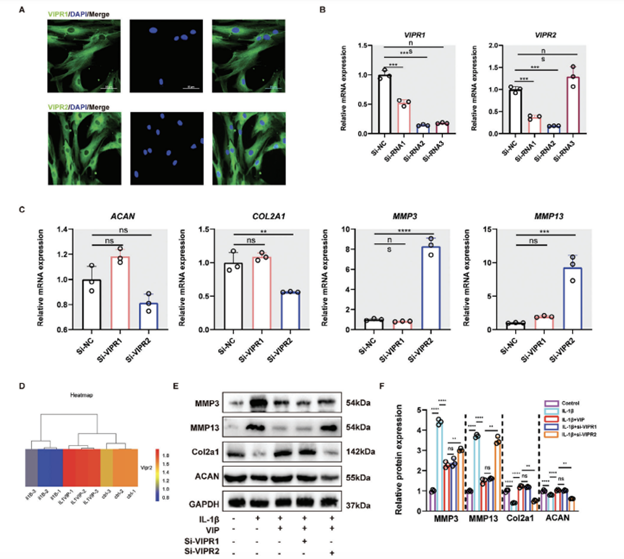
Figure 2. VIP protected IL-1B-induced human NP cells degeneration mainly via VIPR2. A) Representative images of IF staining for VIPR1 and VIPR2 in normal human NP cells. B) qRTPCR results of VIPR1 and VIPR2 of human NP cells in different groups as well as the siRNA sequence with the highest knocking-down efficiency. C) qRT-PCR results of ACAN, COL2A1, MMP3/13, TNFa IL-1B, Bax, and BCL-2 of human NP cells in si-NC, si-VIPR1, and si-VIPR2 group, respectively. D) mRNA-seq results of VIPR2 of human NP cells in control, IL-1B treated, and IL-1B-VIP treated groups, respectively. E) Representative western blotting images of MMP13, COL2A1, and ACAN of human NP cells in Control, IL-1B, IL-1B+VIP, IL-1B+VIP+si-NC, si-VIPR1, and IL-1B+VIP+si-VIPR2 group, respectively. F) Quantification of Western blot results. All data are shown as the mean ± SD. Significance was calculated by Student’s t-test. *p < 0.05, **p < 0.01, ***p <0.001, ****p < 0.0001. ns: Not significant.
VIP Protected Human NP Cells against Inflammation-Induced Apoptosis
Our prior research showed that vasoactive intestinal peptide (VIP) could enhance the viability of human NP cells and improve the expression of ECM-related genes, particularly at a concentration of 10^−8 M (Figure S2, Supporting Information). Nonetheless, VIP’s precise impact on human NP cells treated with inflammation remains to be clarified. Flow cytometry results indicated that treatment with IL-1β for 24 hours significantly doubled the apoptosis rate of human NP cells. Conversely, VIP treatment notably reduced the increase in apoptosis, bringing the rate nearly back to baseline levels observed in the control group (Figure 3). This observation was further supported by TUNEL staining (Figures 3B,C).
The impact of VIP on the expression of pro-apoptotic proteins was also assessed. Immunofluorescence (IF) staining was used to examine cleaved-caspase 3, a protein associated with apoptosis. The findings revealed that VIP markedly decreased the IL-1β-induced expression of cleaved-caspase 3 in human NP cells (Figures 3D,E). Thus, VIP appears to provide protective effects against IVDD by mitigating inflammation-induced apoptosis in NP cells.
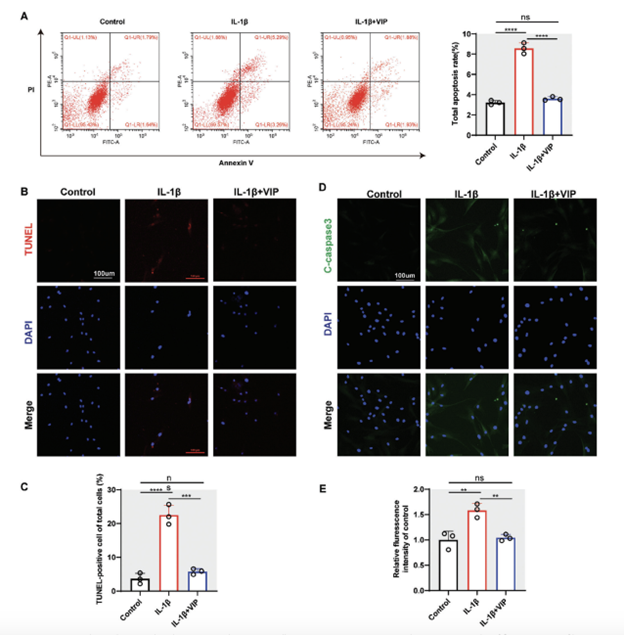
Figure 3. VIP ameliorated IL-1B-induced apoptosis in human NP cells. A) Representative images and quantitative results of flow cytometry of human NP cells in control, IL-1B treated, and IL-1B-VIP treated groups, respectively. B,C) Representative images and quantitative results of TUNEL staining of huma ion of the ratio of TUNEL-positive NP cells. D,E) Representative images and quantitative results of IF staining for cleaved-caspase 3 of human NP cells in control, IL-1B treated, and IL-1B -VIP treated groups, respectively. All data are shown as the mean± SD. Statistical significance was determined by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns: Not significant.
VIP Alleviated Inflammation-Triggered Excretion of Pro-Inflammatory Factors and ECM Degradation in Human NP Cells
“Excessive inflammation is the critical pathology factor during IVDD. Gene expression quantitative analysis revealed that VIP could dose-dependently alleviate IL-1B-induced expression of pro-inflammation-related gene, including NOS2, TNFa, apoptosis-associated speck-like protein (ASC), and IL-1B (Figure S2, Supporting Information). IF staining demonstrated that NP cells treated by IL-1B had increased the protein expression of iNOS, whereas the expression of iNOS was remarkably suppressed with the pre-treatment by VIP (Figure 4A,B). In addition, we evaluated the effect of VIP on ECM homeostasis within inflammatory environment. The results of IF staining revealed that IL-1B evidently promoted the gene expression of MMP3 but repressed the gene expression of ACAN, whereas VIP partly alleviated these abnormal effects (Figure 4C,D). Notably, Western blot suggested consistent results with the results above (Figure 4E,F). Collectively, VIP could alleviate inflammation-triggered excretion of pro-inflammatory factors and ECM degradation in human NP cells.”
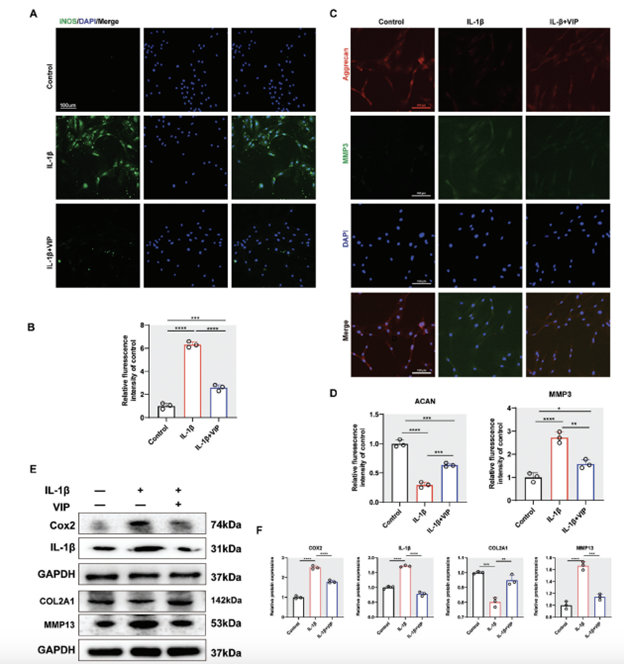
Figure 4. VIP alleviated IL-1B-induced inflammatory response and ECM disequilibrium in human NP cells. A) Representative images of IF staining for iNOS of human NP cells in control, IL-1B treated, and IL-1B-VIP (10−8 m) treated groups, respectively. B) Quantification of the expression of iNOS. C) Representative images of IF staining for aggrecan and MMP3 of human NP cells in control, IL-1B treated, and IL-1B-VIP treated groups, respectively. D) Quantification of the expression of aggrecan and MMP3. E) Representative western blotting images of COX2, apoptosis-associated speck-like protein (ASC), IL-1B, COL2A1, and MMP13 in human NP cells in control, IL-1B (10 ng mL−1) treated, and IL-1B-VIP (10−8 m) treated groups, respectively. F) Quantification of Western blot results. All data are shown as the mean ± SD. Statistical significance was determined by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns: not significant.
Activation of AKT Signaling Pathway Mediated the Protective Effects of VIP in Inflammation-Induced NP Cells Degeneration
In order to identify the key signaling pathways underlying the protective effects of VIP on NP cells, we conducted bulk transcriptome sequencing on human NP cells treated under various conditions in vitro. The data, depicted in Figure S3 (Supporting Information), showed that IL-1β treatment led to the upregulation of 687 genes and downregulation of 526 genes. In contrast, NP cells that had been pre-treated with VIP exhibited fewer changes, with 179 genes upregulated and 74 genes downregulated (Figure 3A, Supporting Information). The distribution of these differentially expressed genes is visually represented in a heatmap (Figure 5A), indicating that VIP treatment mitigated the aberrant gene expression induced by IL-1β in NP cells.
Further analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pinpointed the PI3K-AKT signaling pathway as a potential mediator of VIP’s protective actions against the degeneration of NP cells caused by IL-1β (Figure 5B). Gene Set Enrichment Analysis (GSEA) also supported the role of VIP in influencing IL-1β-treated NP cells (Figure 3B, Supporting Information). To verify the significance of the PI3K-AKT pathway in the regulation of VIP’s protective effects against inflammatory degeneration in NP cells, we executed Western blot analyses. These analyses indicated that VIP notably increased the phosphorylation levels of AKT compared to cells treated solely with IL-1β (Figure 5C). However, this effect was significantly hindered by the AKT inhibitor LY294002 (Figure 5C).
Immunofluorescence (IF) staining yielded results consistent with the Western blot findings regarding AKT (Figures 5D,F). As anticipated, treatment with VIP increased the protein levels of ACAN, COL2A1, and BCL-2, while reducing the expression of COX2 in NP cells affected by IL-1β. This effect was reversed by the AKT inhibitor (Figures 5G,H). Thus, the activation of the AKT signaling pathway appears to be crucial for mediating the protective effects of VIP in NP cells undergoing inflammation-induced degeneration.
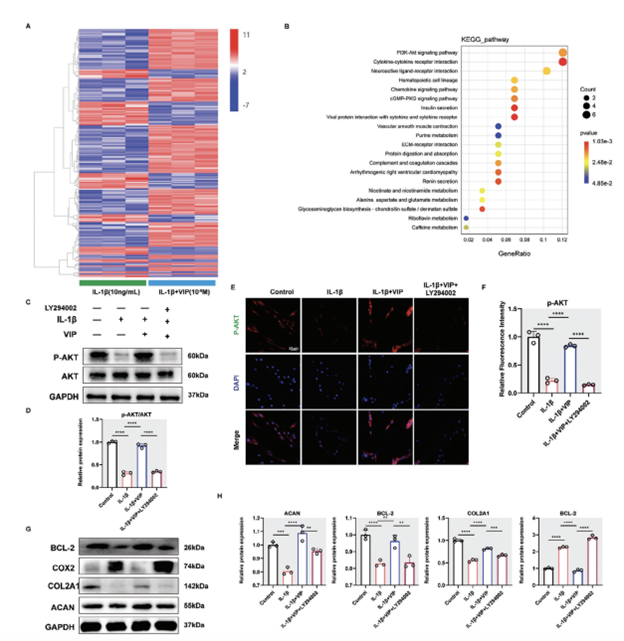
Figure 5. VIP regulated IL-1B-induced human NP cells degeneration by activating PI3K/AKT signaling pathway. A) Heatmap depicting all abnormally expressed genes in human NP cells from control, IL-1B group, and IL-1B+VIP group. B) KEGG analysis showed the top 20 differentially signaling pathways in human NP cells from IL-1B group and IL-1B+VIP group. C) Representative western blotting images of p-AKT and AKT in human NP cells from Control, IL-1B (10 ng mL−1), IL-1B+VIP (10−8 m), and IL-1B+VIP+PI3K inhibitor (LY294002, 0.1um) group, respectively. D) Quantification of Western blot results. E,F) Representative images and quantification results of IF staining for p-AKT in human NP cells from Control, IL-1B, IL-1B+VIP, and IL-1B+VIP+ LY294002 groups, respectively. G,H) Representative western blotting images of BCL-2, COX2, COL2A1, and ACAN in human NP cells from Control, IL-1B (10 ng mL−1), IL-1B+VIP (10−8 m), and IL-1B+VIP+PI3K inhibitor (LY294002, 0.1um) group, respectively. All data are shown as the mean ± SD. Statistical significance was determined by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns: not significant.
Identification of FGF18-FGFR2 as the Bridge Between VIP and PI3K/AKT Signaling Pathway
To delve deeper into the molecular mechanisms by which VIP/VIPR2 activates the AKT signaling pathway, we revisited the differentially expressed genes associated with this pathway. Among them, FGFR2, CSF1, PIK3R5, THBS2, and Tnn were identified as related to the AKT pathway. Our qRT-PCR findings indicated that FGFR2 was the only gene showing significant differential expression across various NP cell groups (Figure 6A). Subsequent Western blot analysis suggested FGFR2 as a potential intermediary that facilitates the interaction between VIP and the AKT signaling pathway (Figure 6B).
We further explored FGFR2’s role in human NP cells by introducing a specific si-FGFR2 system. Following the suppression of FGFR2, there was a notable decrease in the expression of ACAN, COL2A1, and BCL-2, while the expression levels of MMP13, Bax, and IL-1β increased, highlighting FGFR2’s essential role in maintaining NP cell homeostasis (Figure 6D). Similar outcomes were observed when FGFR2 was inhibited in NP cells using Alofanib, confirming these findings (Figure S4, Supporting Information). Crucially, VIP treatment alleviated the negative impacts of IL-1β on the AKT pathway; however, this protective effect was nullified by the knockdown of FGFR2 (Figure 6E).
It has been previously established that the binding of FGF to FGFR leads to the activation of FRS2 and the PI3K/AKT signaling cascade through subsequent interactions. In our study, we noted a reduction in p-FRS2 protein levels in NP cells following IL-1β stimulation, which was partially reversed by VIP addition (Figure 6E; Figure S7A,B, Supporting Information). IF staining demonstrated that VIP could counteract IL-1β-induced ECM degradation, but this was significantly hindered when FGFR2 expression was suppressed (Figures 6F,G), linking the protective effects of VIP on IL-1β-induced degeneration in NP cells to FGFR2 activity.
Our investigation also covered FGF-related genes, with FGF18 standing out due to its consistent pattern with FGFR2 (Figure S5A, Supporting Information). STRING analysis confirmed a direct interaction between FGF18 and FGFR2 (Figure S5B, Supporting Information). Notably, qRT-PCR results showed an increase in FGF18 levels in the IL-1β+VIP group compared to the IL-1β group alone (Figure S5C, Supporting Information). Additionally, introducing FGF18 to human NP cells mitigated ECM degradation, echoing previous findings with chondrocytes (Figure S5D, Supporting Information).
Collectively, these results suggest that the FGF18-FGFR2 axis plays a crucial role in mediating the protective effects of VIP on IL-1β-treated human NP cells through the AKT signaling pathway.
VIP/VIPR2 Regulated the Expression of FGF18 via miR-15a-5p
Considering that the VIP/VIPR2 pathway modulates FGF18 at the mRNA level, we delved into identifying a potential intermediary between VIP/VIPR2 and FGF18. MicroRNAs (miRNAs), which are small non-coding RNAs measuring 22–25 nucleotides in length, play a significant role in disease development, including intervertebral disc degeneration, by directly influencing the mRNA levels of specific genes. To uncover the underlying regulatory mechanism of VIP/VIPR2 on FGF18 expression, we initially searched for potential bridging miRNAs targeting FGF18. This was achieved using computational tools such as Target Scan (Target Scan Human 7.2), mirDIP, and miRDB. These databases collectively identified 12 miRNAs potentially targeting the 3′-UTR of FGF18 mRNA (Figure S6A, Supporting Information).
Subsequently, we categorized nucleus pulposus (NP) cells into three groups: control, IL-1β-treated, and IL-1β + VIP-treated. RT-qPCR analyses were conducted, revealing that miR-15a-5p levels increased significantly following IL-1β treatment and decreased after VIP administration. This pattern suggests that miR-15a-5p could be the intermediary molecule mediating the effects of VIP/VIPR2 on FGF18/FGFR2 signaling (Figure S6B, Supporting Information).
Further validation was conducted through a luciferase reporter assay to determine if miR-15a-5p directly targets FGF18 by binding to its 3′-UTR in NP cells stimulated by IL-1β. The assay results displayed a notable reduction in luciferase activity in NP cells co-transfected with miR-15a-5p mimic and wild-type (WT) FGF18 compared to the control group, indicating direct interaction (Figure S6C, Supporting Information). No significant change in luciferase activity was observed in the mutant (MUT) FGF18 group regardless of miR-15a-5p mimic or miR-NC transfection (Figure S6D, Supporting Information). Western blot results further confirmed the suppressive effect of miR-15a-5p on FGF18 expression in NP cells, establishing FGF18 as a direct target of miR-15a-5p (Figures S6E–H, Supporting Information). Notably, the miR-15a-5p binding sequence within the 3′UTR of the FGF18 gene is conserved across several species, highlighting the structural basis for targeting FGF18 as a therapeutic strategy for IVDD treatment.
In summary, the VIP-VIPR2 axis influences FFG18/FGFR2 signaling by increasing the mRNA levels of FGF18 through the mediation of miR-15a-5p.
VIP Alleviated Lumbar Instability-Induced Intervertebral Disc Degeneration by Modulating FGFR2/FRS/AKT Pathways in Vivo
“To verify that VIP delays IVDD in vivo, we administrated exogenous VIP to mice with lumbar instability-induced lumbar IVDD via a sustained release pump (1004 W, RWD, Shenzhen, China) for continuous 4 weeks. The VIP-treated group’s Pfirrmann grade scores were significantly lower than those of the IVDD group according to MRI four weeks after surgery (Figure 7A,B). VIP could slow the progression of lumbar instability-induced IVDD in vivo. Consistently, histological analysis demonstrated a relatively vague frontier between NP tissue and AF tissue, as well as evidently reduced NP tissue and glycosaminoglycan in IVDD group. However, in VIP-treated group, there was still a little part of NP tissue within pathological IVD (Figure 7C–E). Likewise, IF staining unveiled that the expression of ACAN was partly reversed in VIP-treated group, accompanied by less expression of MMP3, compared with the IVDD group (Figure 7F,G). In addition, IHC staining in VIP-treated group, the expression of phosphorated FRS2 was also elevated (Figure 7C). Overall, these results indicated the therapeutical potential of VIP addition to ameliorate IVDD.”

Figure 7. VIP alleviated lumbar instability-induced intervertebral disc degeneration by modulating FGFR2/FRS/AKT pathways in vivo A) Representative MRI images of mice lumbar spine in control, IVDD, and IVDD+VIP groups. B) Degenerated score of lumbar IVD in mice model based on T2-weighed MRI. C) Representative images of H&E staining for lumbar IVD in mice model from control, IVDD, and IVDD+VIP groups. D) Representative images of SOFG staining for lumbar IVD in mice model from control, IVDD, and IVDD+VIP groups. E) Histological score of lumbar IVD in mice model from control, IVDD, and IVDD+VIP groups. F,G) Representative images of IF staining and quantification results for aggrecan and MMP3 of lumbar IVD in mice model from control, IVDD, and IVDD+VIP groups. All data are shown as the mean ± SD. Statistical significance was determined by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns: not significant.
Discussion
“The etiology of IVDD is multifactorial disorders. Unfortunately, in the current state of research, there are no effective drugs that can delay the progression of IVDD. In the past few decades, various types of nerve fibers, including sensory and sympathetic nerve fibers, have been observed within degenerated NP tissue, and the focus was mainly centered on IVDDtriggered LBP.
However, research regarding the role of nerve systems and their secreted neuropeptides on the function of NP cells remains limited. VIP plays vital roles in various biological functions, and its anti-inflammatory and immune-modulatory ability has been recognized as a potent candidate for treating inflammation-related diseases.
In this present study, we first systematically evaluated the expression of VIP system within human NP tissue. We found that the expression of VIPR1 and VIPR2 correlated negatively the severity of IVDD. Inflammation or oxidative stress can decrease the gene expression of VIPR1 and VIPR2. We also found that VIP showed efficiently protective effects on IL-1B-induced cell dysfunction and ECM degradation in human NP cells and that the activation of AKT signaling pathway may be involve in VIP/VIPR2 axis-mediated protection of NP cells. Additionally, this study also revealed FGF18/FGFR2 as the bridge protein between VIP/VIPR2 and AKT signaling pathway. Further, VIP-VIPR2 axis regulated FFG18/FGFR2 signaling pathway via enhancing the mRNA level of FGF18 mediated by miR-15a-5p.
Finally, via lumbar instability-IVDD mice model, we confirmed the therapeutical effects of VIP in delaying IVDD via modulating FGFR2/FRS/AKT pathways in vivo. NP cells apoptosis is considered as the major mechanism for decreased cell numbers within NP tissue during IVDD. Abnormal NP tissue can further contribute to changes of biomechanics and following low back pain. In this present study, we revealed that VIP could efficiently reduce IL-1B-induced NP cells apoptosis. The results above demonstrated the antiapoptotic effect of VIP on inflammation-treated NP cells. OA is also a chronic and degenerative disease resulting from dysfunction of chondrocytes. The pathological of OA is similar to IVDD, and both diseases display cell apoptosis, senescence, and ECM degradation. Previous evidence has suggested that VIP could prevent chronic cartilage damage and joint remodeling in an experimental animal model. Excessive activation of inflammatory response is another typical feature for IVDD. Previous study reported various proinflammatory cytokines related to IVDD.Among these pathological mediators, IL-1B and TNFa have been the most studied. These two cytokines have been used to establish IVDD model in vivo or in vitro. Therefore, in this study, we used IL-1B as the mediator to induce NP cells degeneration model in vitro. As shown in results, in addition to increasing cell apoptosis and senescence, IL-1B significantly promoted the gene and protein expression of inflammation-related molecules. In addition, ECM anabolism-related molecules were evidently downregulated by IL-1B. Nevertheless, addition of exogenous VIP can efficiently restore these deleterious events. In fact, VIP showed various anti-inflammatory role in previous studies. Sun et al. found that Lentivirus expressing VIP inhibited inflammation in mice with acute lung injury induced by LPS in vivo. Similarly, Liang et al. revealed that overexpression of VIP in rat knee joint could efficiently alleviate the progression of OA by suppressing the production of inflammation- and ECM-related cytokines. Therefore, the results of this study increased the indication of VIP in anti-inflammatory effects, and provided new treatment target for IVDD.
Different signaling pathways will be activated or inhibited during IVDD. In this study, we performed bulk transcriptome sequencing analysis, and found that PI3K-AKT signaling pathway may the critical pathway to mediate VIP’s protecting NP cells against inflammatory damage. In addition, this study also revealed that knocking down VIPR2 resulted in more evident blocking effects on VIP-mediated NP cells protection than VIPR1, suggesting that VIP possibly exerted its protective role in delaying IVDD mainly via VIPR2. To further uncover the underlying mechanism between VIP/VIPR2 axis and PI3K/AKT axis, we re-analyzed the differentially expressed related to AKT signaling pathway, and found that FGFR2 showed the most protein interaction score. FGF signaling has been widely reported to participate in procession of OA mainly via FGFR1-3. The deletion of FGFR1 in chondrocytes can delay cartilage degeneration in aged mice, whereas FGFR3 has been shown to prevent progression of osteoarthritis in the knees and jaws. Nevertheless, in this study, we found that FGFR2 was indispensable for maintaining the homeostasis of NP cells. In fact, previous study also has identified FGFR2 as a novel specific biomarker for NP cells.
Interestingly, we found that the interaction of FGF18 and FGFR2 may be the critical phase in the activation of AKT signaling. Notably, addition of FGF18 to human NP cells could also showed protective effects on ECM homeostasis, which was similar to a previous study with the finding that It was demonstrated that FGF18 could stimulate the proliferation and accumulation of type II collagen and proteoglycans in cultured porcine articular chondrocytes, as well as in adult human articular chondrocytes. However, silence of FGFR2 blocked the phosphorylation of FRS2 and AKT induced by VIP. Taken together, we deduced that FGF18/FGFR2 axis may bridge the connecting between VIP/VIPR2 and FRS2/AKT signaling pathway. Establishment of IVDD model has been the essential step for studying the therapeutical effects of VIP in delaying IVDD. In this study, we combined MRI, H&E staining, IF staining, and IHC staining to determine the effects of VIP in vivo. The results were almost consistent with in vitro experiments, which suggested that VIP might delay the procession of IVDD by promoting expression of FGF18-FGFR2, thereby activating PI3K/AKT signaling pathway. Notably, the effects of VIP on adjacent healthy segments were not evaluated in this study, which will add more information in VIP-IVD axis in future study. Therefore, we concluded that VIP-VIPR2 showed protective effects of inflammation-induced IVDD via activating AKT signaling pathway, and this signal axis was achieved by means of FGF18- FGFR2-FRS2 cascades. In addition, VIP-VIPR2 axis regulated FFG18/FGFR2 signaling pathway via enhancing the mRNA level of FGF18 mediated by miR-15a-5p (Figure 8).”
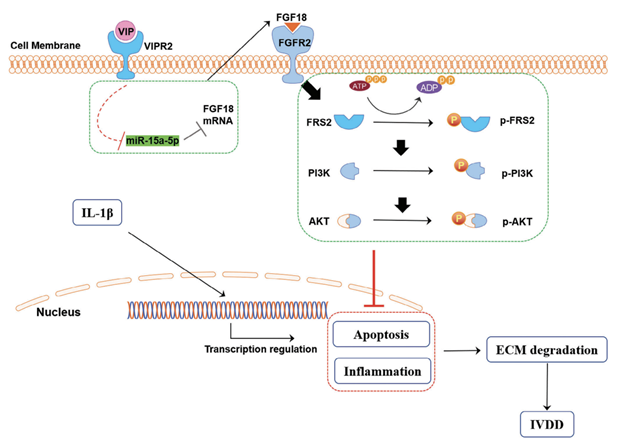
Figure 8. The mechanism by which VIP regulated the FGF18/FGFR2-PI3K/AKT signaling pathway in IVDD Sympathetic neurotransmitter, VIP, bind its receptor, VIPR2, and increased the mRNA expression of FGF18 via suppressing the expression of miR-15a-5p. Subsequently, increased connection between FGF18 protein and FGFR2 protein would successively activate FRS2, PI3K, and AKT. Then, enhanced AKT signaling pathway significantly alleviated inflammation-triggered cell apoptosis, inflammation, and ECM imbalance in human NP cells, and delayed IVDD.
Conclusion
In conclusion, this present study not only revealed the expression changes of VIP-VIPRs system within human NP tissue, but also demonstrated the protective effects of VIP on inflammation mediated NP cells degeneration. The present study highlights the potential of VIP in clinical transformation, particularly in the treatment of IVDD in the future.